


Description
Specific, unbiased rRNA depletion and library prep for RNA-Seq.

RNA samples frequently contain up to 90% ribosomal RNA. Transcriptome sequencing without removing these overabundant rRNA sequences reduces sequencing capacity and makes it harder to detect lower expressing, biologically relevant transcripts. See how CRISPRclean can help your research with effective rRNA removal.
Highlights
- Effective single tube, multi-species depletion with CRISPR-Cas9 mediated technology
- Increases sensitivity to detect lower expressing, biologically relevant transcripts
- Optimized library prep and depletion workflow generates high quality representation of transcripts
- Consistent full-length, uniform transcript coverage, and high library complexity
- One day workflow: 7.5 hours assay with 3 hours of hands-on time
| Specification | |
| Catalog | JUM-KIT1014 |
|---|---|
| Samples per kit | 24 |
| Assay time | 7.5 hours |
| Hands-on time | 3 hours |
| Sample types | Total RNA |
| Input quantity | 5 to 100 ng |
| Strand specificity | >98% Directional and strand specific |
| Compatible species | Human, mouse, and rat |
| Designed to deplete | Human 5S, 5.8S, 18S and 28S rRNA genes, 45S rRNA precursor, mitochondrial 12S and 16S rRNA genes |
| Multiplex | Up to 96 unique dual index adapter barcodes. Requires KIT1017 CRISPRclean Unique Dual Index Adapter Plate for RNA Prep (Set A) |
One-day workflow from RNA to sequencing-ready library


Specific and unbiased
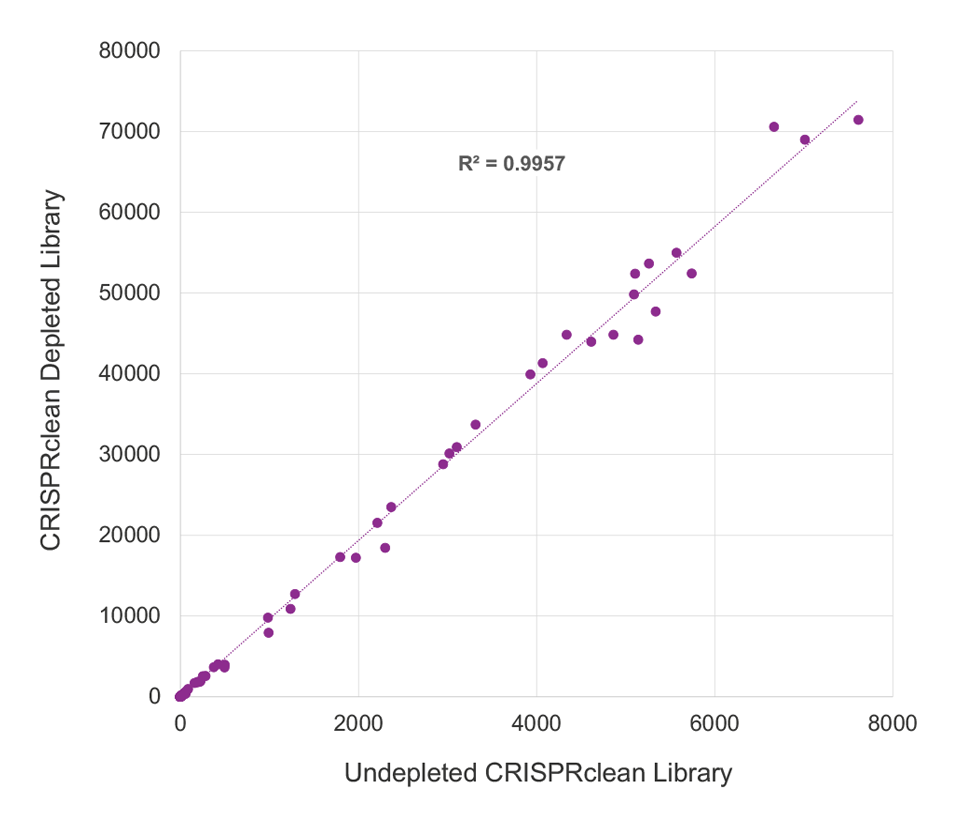
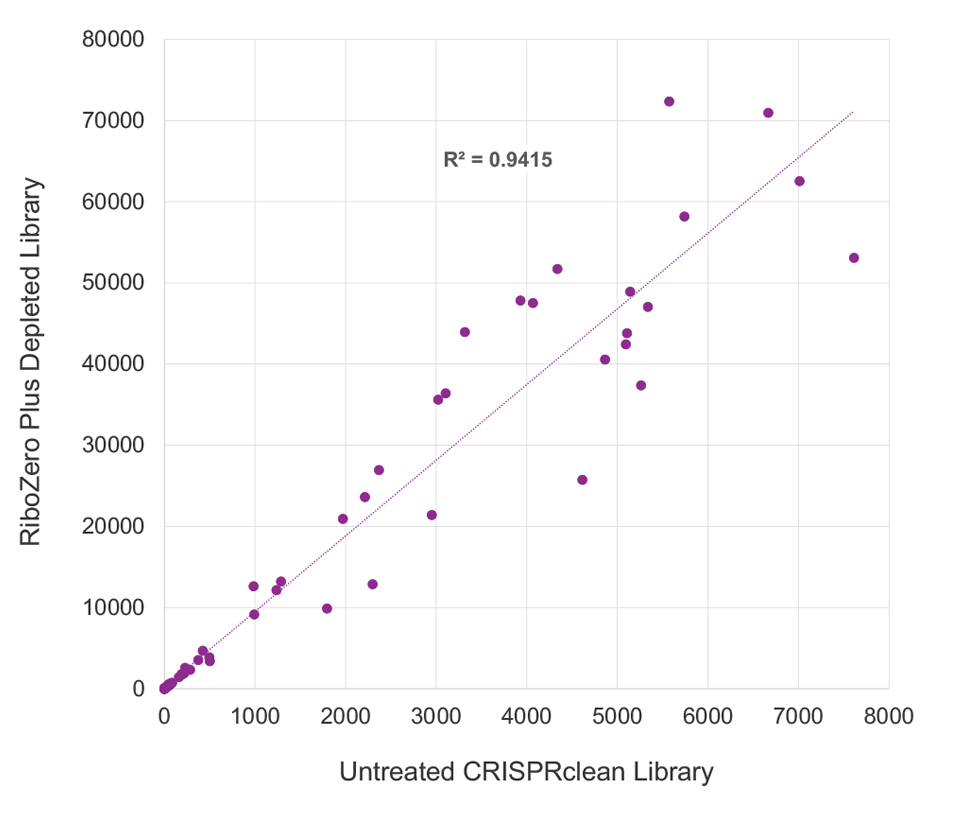
CRISPRclean Stranded Total RNA Prep with rRNA Depletion produces extremely low library bias. ERCC reads counts were highly correlated, between depleted (y-axis) and undepleted libraries (x-axis), indicating the CRISPRclean rRNA depletion method is highly specific, unbiased, and accurate. This allows for gene expression measurements to be more accurately represented than those that would be obtained from an undepleted sample. Illumina RiboZero Plus Depletion paired with Jumpcode’s stranded RNA prep shows lower ERCC read count correlation and higher bias.
CRISPRclean Depletion
|
RiboZero Plus Depletion
|
Effective single tube, multi-species depletion

>90% rRNA depletion across mammalian species. Universal reference RNAs (Human, mouse, rat, chicken, cow, and dog) were used to assess performance of the ribodepletion across multiple species.
Optimized library prep and depletion workflow

High quality representation of human gene expression

Consistent full-length, uniform transcript coverage

Increases sensitivity to detect lower-expressing, biologically relevant transcripts
